Fernando Garcia
Home Research Blog Other About me
Some formalizations in thermal theory
March 10th, 2024.
Introduction
Thermodynamics is a topic that's highly tied to experiments. It is pretty much the norm that books and courses on thermodynamics follow a historical treatment with a big emphasis on measurements and experiments in general. The introduction of new concepts comes with a related experiment or lab scenario.
While studying some thermodynamics on my free time, I came across a new presentation that brings more rigor to those that are less experiment-driven. In this post I want to outline this methodology and derive some of the results that are familiar from a course in thermodynamics.
This post was inspired by the works of László Tisza and the book Thermal Quantum Field Theory: Algebraic Aspects and applications, by Khanna, Malbouisson, Malbouisson, and Santana.
Although previous knowledge is not necessarily required to follow the work below, it is highly recommended.
Basic notions
Here's a list of variables and the letters we shall use to denote them:
Variables can be of two types: Extensive and Intensive. Intensive properties are those that are independent of the size of the system. On the other hand, extensive variables are those that depend on the size of the system. As an example, consider a blob of clay:
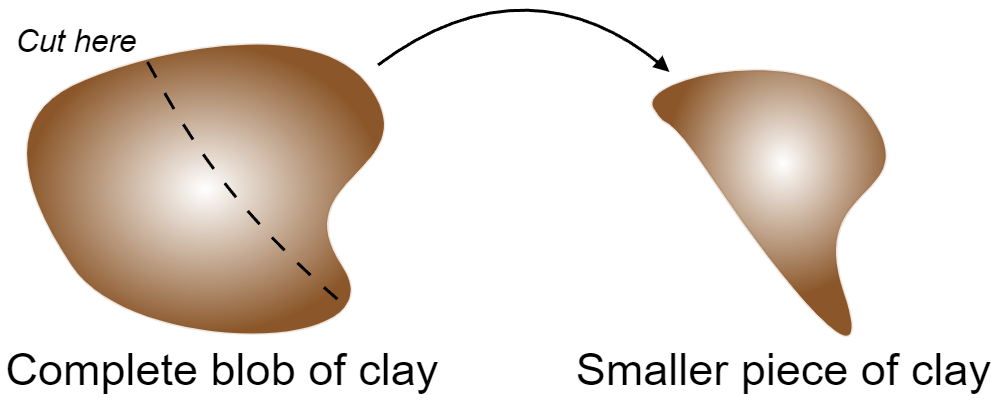
The new piece of clay will have the same temperature and (mass) density, making them intensive variables. Still, it will have different energy, volume, and number of moles, making them extensive variables.
There are two important remarks to be made here: Densities of extensive variables are intensive, and there are variables which are neither intensive or extensive.
In the same way we can define a system's state in classical mechanics by describing its positions and momentums, we define a thermal system's state by referring to its thermal variables. In the same way we are interested in knowing the equations of motion in classical mechanics, given the classical variables of position and momentum, our goal in thermal physics is to have a function of state for the system in question.
Before moving into a brief summary of the three laws of thermodynamics, we need to make sure two points are clear: Processes are assumed to be quasi-static, meaning that states are connected by an infinite amount of intermediary states. Further, suppose we describe a system with
We will set
Where the
With this in mind, we can talk about the three laws. These allow us to talk about the dynamics, rather than just specific states of a system.
1st law: Conservation of energy. The (infinitesimal) change in internal energy is equal to a flow in heat and work:
2nd law: On entropy and the (ir)reversibility of process. We introduce, as an assumption, the state function
Extensive variables (given the restrictions imposed by internal constraints) will take values which maximize
There are processes which we do not observe in nature and which would not violate the first law. They don't happen because of the second law.
3rd law: It is impossible to lower the temperature of a system to
The fundamental relation
Since
Those two relations are known as the fundamental equations/relations. As we shall see below, we can derive everything from them. Further, we can add a further layer of abstraction and consider the fundamental equation:
And talk about the entropy representation:
Or the energy representation:
Both (internal) energy and entropy are first-order homogeneous functions:
So we have in general
This might seem like a trivial remark, but it is a useful one: Differentiate the above expression with respect to the parameter
But we can also take the differential of
Where
This means that if we know the
What can we say about the
Since
With this in mind, consider taking
Meaning that the
To see how the
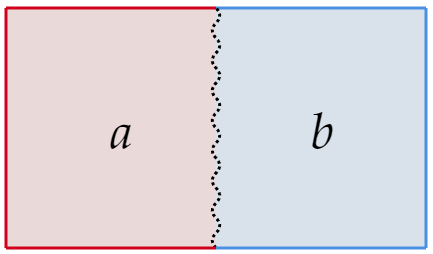
With total energy
And total entropy
For this first example, let's keep
Equilibrium dictates
So
Or in terms of
Where
If we move out of equilibrium, what happens to the system? The evolution will be characterized by
There are two cases to consider:
Case 1:
Case 2:
It is clear that heat will travel from the low
So
In general
In particular, notice two important things: The first being that
Since
As another example, consider a simple system, meaning that
Where
Since
Meaning
If we move out of equilibrium, what happens to the system? The evolution will be characterized by
There are two cases to consider:
Case 1:
Case 2:
It is clear that the chamber that will grow (positive change in volume) is the one with higher "force"
From definitions and as above, we have:
So we identity
In similar ways, we find that
Which allow us to write (for the simple system) in the Energy representation:
And in the entropy representation:
Using the first law (
Thermodynamic potentials
Let's now consider the Legendre transform of
With
What this does is create a function
And we refer to the
Consider a simple system in the energy representation. Then
1. Helmholtz free energy.
Take the Legendre transform in the variable
2. Enthalpy
Take the Legendre transform in the variable
3. Gibbs free energy
Take the Legendre transform in the variable
4. Grand thermodynamical potential (also known as Landau potential or Landau free energy)
Take the Legendre transform in the variable
Consider now some transforms in the Entropy representation:
5. Massieu potential (also known as Helmholtz free entropy)
Take the Legendre transform in the variable
6. Planck potential (also known as Gibbs free entropy)
Take the Legendre transform in the variable
These ideas provide some advantages, such as the description of a thermal state in terms of both intensive and extensive variables.
Final remarks
As usual, the post is getting long. In a later blog post I will discuss further developments in the aforementioned way of doing thermodynamics. There is relevance in Statistical mechanics, a topic I have yet to discuss here.